近日,来自广州医科大学附属第五医院前沿医学交叉研究中心、天津大学生物信息中心、国家人类基因组北方研究中心、南新制药等单位的研究团队合作在病毒分类研究中取得新进展,研究成果在生物信息学领域顶级期刊Briefings in Bioinformatics(中科院分区一区Top期刊,影响因子11.6)上发表。
如何对病毒群体进行准确而客观的分类对于病毒防控至关重要,目前常用的两种方法,分别是基于单核苷酸多态性(SNP)和系统发育树来对病毒亚群进行分类。基于SNP的分类比较简单直接,适用于小范围流行爆发的病毒(如埃博拉病毒)、进化缓慢的病毒(如约翰·坎宁安病毒)或进化迅速但谱系更替率低的病毒(如丙型肝炎病毒),然而该方法难以充分揭示复杂流行病毒的遗传多样性。相比之下,基于系统发育树的分类能提供较详细的病毒谱系,但其严重依赖于人为设定的遗传距离阈值来定义密切相关病毒亚群间的最大遗传差异。尽管也可基于统计学标准来定义遗传距离阈值,但具有统计不确定性。此外,用于构建系统发育树的多序列比对结果需要所有序列保持对位排列,这将丢失大量有用信息。同时,由于计算时间和内存消耗过大,很难同时将成千上万条基因组序列进行排列并构建超大的系统发育树。PhenoGraph是近年来开发的一种基于无监督机器学习的算法,它能解析高维数据的内部自然结构,其优越的计算能力可对上百万条病毒序列进行详细分类而不至于消耗过多的时间或内存。该方法最初旨在从高维单细胞数据中解析功能不同的细胞亚群,分类精确率高达99.85%。虽然PhenoGraph是一种性能优异的分类方法,但其主要用于单细胞测序数据的细胞分类,因此,如何将该方法应用到病毒分类领域是一个值得探索的问题。
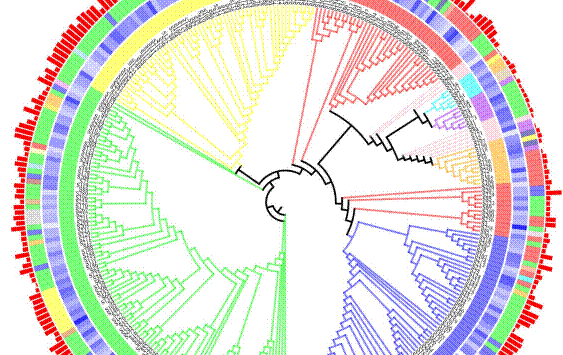
在本工作中,研究团队以一种广泛流行病毒的基因组序列为研究对象,从近二十万个病毒序列中检出其全部突变信息,发现该病毒基因组的GC含量随时间的推移而持续降低,这可能有利于病毒传播,并发现其移码突变十分罕见。接下来研究人员以二进制的方式对所有基因组突变进行编码,一方面可以最大限度地避免用于分类的遗传信息的丢失,另一方面该二进制编码的遗传信息能较好地适用于PhenoGraph分类。通过采用PhenoGraph对该二进制编码的病毒数据进行分类成功鉴定出303个病毒亚群。经比较发现,基于PhenoGraph的分类结果与已知的GISAID分类(S, L, V, G, GH, GR, GV和O)基本一致,但更为详细和精确。基于变化趋势分析发现该病毒遗传多样性的增长速度已明显放缓。此外,该团队还分析了各亚群之间的时间、空间和系统发育关系,在一定程度上揭示了该病毒的进化轨迹。因此,本研究工作不仅有助于更好地理解复杂流行病毒基因组进化及其流行病学模式和趋势,也为疾病的防控提供了重要依据。
广州医科大学附属第五医院副教授杨志凯、南新制药潘玲玉、诺赛基因|国家人类基因组北方研究中心副研究员张延明为本文的共同第一作者。广州医科大学附属第五医院副教授杨志凯和天津大学生物信息中心主任高峰教授为本文的共同通讯作者。广州医科大学附属第五医院为该论文的第一作者单位和通讯作者单位。该项目研究得到了国家重点研发计划项目、广州市重点实验室基金、国家自然科学基金项目以及广州医科大学南山学者人才计划的支持。

病毒亚群的系统发育树。Clade是指在GISAID中定义的八大进化枝;Number是指病毒分离株数的对数值。Country为相应亚群中分离株数量最多的国家。Date对应于每个亚群的最早取样日期,从2019年12月24日至2020年10月05日。


